This document is intended for sponsors of clinical investigations of devices conducted within the scope of the Regulation (EU) 2017/745 (MDR). This document may be supplemented in due course with further questions and answers.
Throughout this document the term ꞌdeviceꞌ is used with the same meaning as in the MDR, i.e., for the purpose of the MDR, medical devices, accessories for medical devices and products listed in Annex XVI of the MDR and to which the MDR applies, shall hereinafter be referred to as ꞌdevicesꞌ.
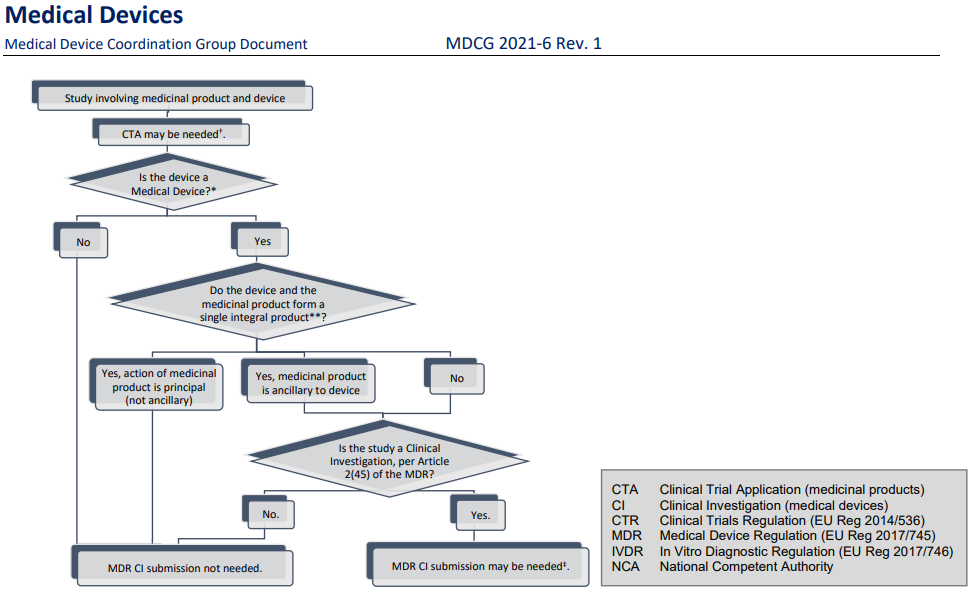
Further, the term “clinical investigation” is used throughout with same meaning as in the MDR Article 2(45), i.e. “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device”.
While clinical investigation is clearly defined in the MDR, it is sometimes also necessary to mention that there are studies that do not fulfil this definition, to draw the line between when the provisions in the MDR chapter VI and annex XV apply, and when they don’t. For this purpose, the broader term “clinical study” is used, and covers, for the purpose of this guidance, studies done within medical research involving humans and includes clinical trials of medicines, clinical investigations of devices, and clinical performance studies of in vitro diagnostic devices, as well as other studies in a clinical setting where products such as drugs or devices are not necessarily involved at all.
Further, the sponsor needs to be aware that MDR does not specify details about ethics review of clinical investigations. It is thus necessary to check national requirements in relation to submission to the Ethics Committee and if applicable, make sure that Ethics Committees and Competent Authorities have access to the same versions of updated documents.
Update - MDCG 2021-6 - Rev.1 - Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation - December 2023
Update - MDCG 2021-6 - Rev.1 - Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation - December 2023
health.ec.europa.eu